A cura di Sebastiano Bianca – Medico Genetista
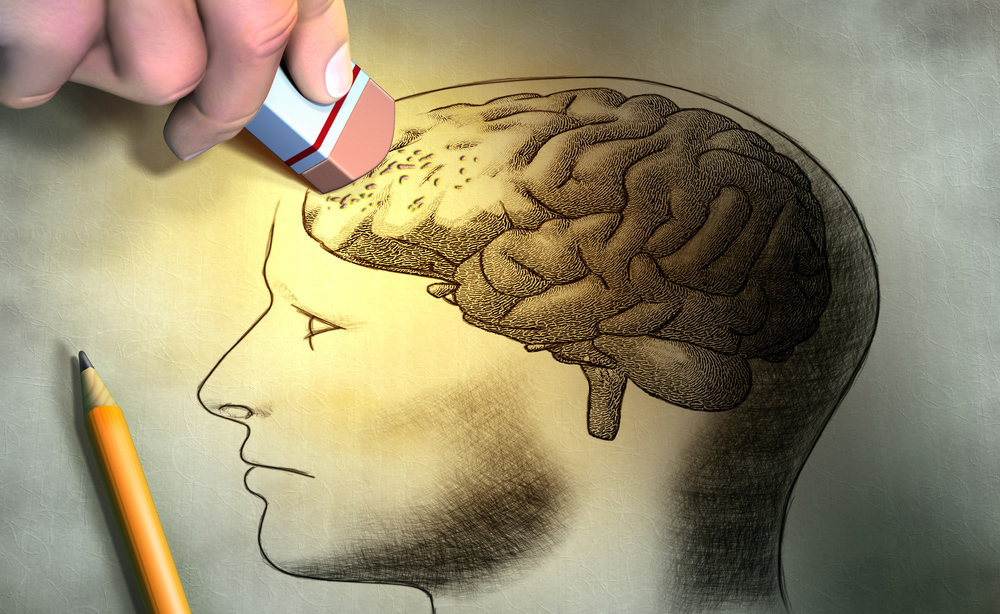
La malattia di Alzheimer è la forma più frequente di demenza neurodegenerativa. La malattia prende il nome da Alois Alzheimer, neurologo tedesco, che nel 1907 descrisse per primo i sintomi e gli aspetti neuropatologici della condizione. La malattia si manifesta con turbe delle funzioni intellettive (memoria a breve termine, orientamento nel tempo e nello spazio, linguaggio, utilizzo degli oggetti, ecc.). Con il tempo si manifesta una progressiva perdita di autonomia con un declino cognitivo che diviene progressivamente sempre più grave.
Aspetti epidemiologici della malattia di Alzheimer
La malattia di Alzheimer si stima interessi circa 24 milioni di persone nel mondo. Il tasso di incidenza annuale della condizione aumenta dall’1% tra le persone di età pari o superiore a 65 anni a circa l’8% per le persone di età pari o superiore a 85 anni. Considerando il previsto invecchiamento della popolazione, queste cifre triplicheranno entro il 2050.
La maggior parte delle forme di Alzheimer sono definite sporadiche, cioè si manifestano come unico caso in una famiglia ed esordiscono generalmente dopo i 65 anni; solo in una minoranza di casi, invece, l’Alzheimer si manifesta in età più giovanile. Circa il 60% di queste forme a esordio precoce sono denominate familiari, ovvero la malattia è presente in più soggetti appartenenti allo stesso nucleo familiare; il 13% di esse è causato dalla presenza di una variante genetica. Si ritiene che la maggior parte dei casi sia causata dall’interazione di diversi fattori di predisposizione genetica con fattori ambientali.
Come si esegue la diagnosi di malattia di Alzheimer?
La diagnosi rimane, a oggi, fondamentalmente clinica. I segni patologici a livello cerebrale risultano caratterizzati dai depositi di proteina β-amiloide extracellulare (Aβ) in placche diffuse e placche contenenti elementi di neuroni degenerati (“placche neuritiche”). I cambiamenti intracellulari sono invece caratterizzati da depositi di proteina tau anormalmente iperfosforilata, ed è inoltre diffusa l’attivazione della microglia e la perdita di neuroni e sinapsi.
Quali sono i geni coinvolti nella malattia di Alzheimer familiare?
Le varianti genetiche a oggi identificate nella malattia di Alzheimer familiare riguardano tre geni denominati presenilina-1 (PSEN1), presenilina-2 (PSEN2) e proteina precursore di beta-amiloide (APP).
Le varianti del gene APP, localizzato sul cromosoma 21, a trasmissione autosomica dominante, rappresentano circa il 14% dei casi di Alzheimer ad esordio precoce, con più di 30 varianti descritte a oggi. Sono state inoltre descritte due varianti a trasmissione recessiva correlate a forme ad esordio precoce. La duplicazione del gene è stata associata alle forme di Alzheimer con angiopatia amiloide cerebrale. Nei soggetti con trisomia 21 la presenza di tre copie del gene APP potrebbe essere alla base della maggiore incidenza di Alzheimer nei soggetti con sindrome di Down. A riprova di questa ipotesi patogenetica si è visto che nei soggetti con trisomia parziale del cromosoma 21, che non include il gene APP, non si manifesta un aumento di casi con Alzheimer.
Altri due geni coinvolti nelle forme familiari di Alzheimer sono i geni PSEN1 e PSEN2 entrambi a trasmissione autosomica dominante. Nel gene PSEN1 sono state descritte ad oggi 185 varianti patogenetiche a trasmissione autosomica dominante, che rappresentano circa l’80% dei casi di Alzheimer familiare a esordio precoce. Infine sembra avere un ruolo anche il gene ADAM10 le cui varianti, riscontrate molto raramente, sono state associate al rischio di malattia ad esordio precoce e familiare sempre con modalità autosomica dominante.
Fattori genetici coinvolti nell’aumento del rischio di insorgenza per le forme ad esordio tardivo
Diversi studi, anche se non sempre concordanti nelle conclusioni, soprattutto in relazione all’etnia della popolazione in studio, hanno attribuito un ruolo come fattore di rischio al polimorfismo ε4 del gene APOE. L’APOE è una proteina che lega i lipidi ed è espressa nell’uomo come tre isoforme comuni codificate da tre alleli, APOE ε2, ε3 e ε4. Un singolo allele APOE-ε4 è associato a un rischio aumentato da 2 a 3 volte, mentre avere due alleli APOE-ε4 si associa ad un aumento del rischio di cinque volte. Sembra inoltre che ciascun allele APOE-ε4 ereditato abbassi l’età di esordio di 6-7 anni. E’ importante comunque sottolineare che APOE è considerato un gene di predisposizione e quindi la presenza di APOE-ε4 non è né necessaria né da sola sufficiente per lo sviluppo della malattia.
Recenti studi mediante sequenziamento genomico hanno individuato altri geni di suscettibilità il cui ruolo necessita di conferma.
In quali individui è indicata l’esecuzione del test genetico
Il test genetico non trova ad oggi un’indicazione nella popolazione priva di fattori di rischio. Risulta indicato nei casi giovanili e nei casi che presentano una storia familiare positiva per la presenza di più soggetti soprattutto se l’insorgenza della condizione si è verificata in età giovanile.
La trasmissione autosomica dominante di molti geni ad oggi coinvolti nell’insorgenza della malattia di Alzheimer fa si che i soggetti portatori di una variante presentino un rischio di trasmissione del 50% per ogni figlio indipendentemente dal sesso. L’esecuzione del test genetico nei familiari sani dei soggetti in cui è stata individuata una variante patogenetica in un gene causativo deve essere necessariamente valutata nell’ambito di una consulenza genetica che illustri le modalità di trasmissione e le possibili implicazioni cliniche e di prevenzione legate all’eventuale riscontro della variante.
Il trattamento e l’utilità del dato genetico
Ad oggi non esiste ancora una terapia per prevenire o guarire l’Alzheimer. I farmaci disponibili migliorano i sintomi e ne rallentano la progressione. Negli ultimi due decenni, i farmaci e i potenziali bersagli farmacologici non si sono tradotti dai modelli animali in terapie efficaci per l’uomo. L’uso di un profilo genetico potrebbe favorire lo sviluppo di farmaci con un approccio terapeutico mirato attraverso l’individuazione di nuovi bersagli terapeutici come le vie del complemento, dello chaperone o del colesterolo. Sono state inoltre identificate varianti in geni che sembrerebbero avere un effetto protettivo riducendo il rischio di malattia. Imitando quindi il meccanismo d’azione di queste varianti genomiche si potrebbero sviluppare nuove terapie più efficaci contro la malattia.
Bibliografia
- Reitz C. Genetic diagnosis and prognosis of Alzheimer’s disease: challenges and opportunities. .Expert Rev Mol Diagn. 2015 Mar;15(3):339-48. (https://pubmed.ncbi.nlm.nih.gov/25634383/)
- Fortea J, Zaman SH, Hartley S, Rafii MS, Head E, Carmona-Iragui M. Alzheimer’s disease associated with Down syndrome: a genetic form of dementia. .Lancet Neurol. 2021 Nov;20(11):930-942. (https://pubmed.ncbi.nlm.nih.gov/34687637/)
- Cruts M, Theuns J, Van Broeckhoven C. Locus- specific mutation databases for neurodegenerative brain diseases. .Hum Mutat. 2012 Sep;33(9):1340-4. (https://pubmed.ncbi.nlm.nih.gov/22581678/)
- J Kuusisto, K Koivisto, K Kervinen, L Mykkänen, E L Helkala, M Vanhanen, T Hänninen, K Pyörälä, Y A Kesäniemi, P Riekkinen, et al. Association of apolipoprotein E phenotypes with late onset Alzheimer’s disease: population based study. BMJ 1994 Sep 10;309(6955):636-8. (https://pubmed.ncbi.nlm.nih.gov/8086986/)




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}