A cura di Cristina Gorgone – Medico Genetista
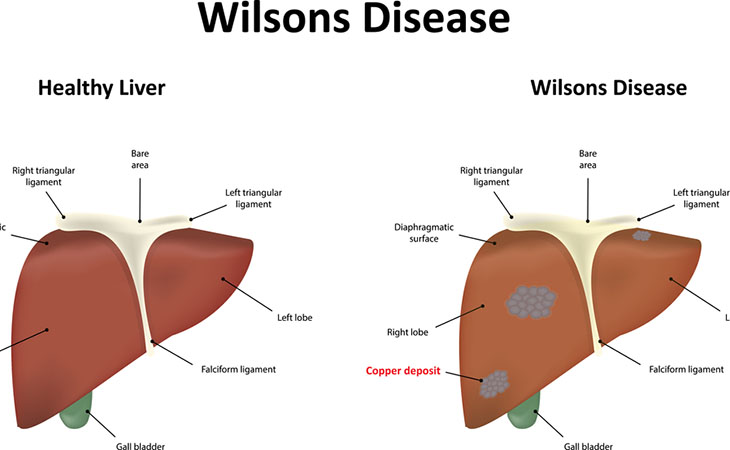
La Malattia di Wilson, descritta per la prima volta nel 1912 dal neurologo americano Kinner Wilson come degenerazione epatolenticolare, è una patologia ereditaria a trasmissione autosomica recessiva dovuta a mutazioni del gene ATP7B. è caratterizzata da un difetto del metabolismo del rame, con conseguente accumulo di rame nei tessuti, specialmente a livello epatico, corneale e cerebrale.
La prevalenza degli individui affetti è di 1:30.000; i portatori hanno una frequenza di 1:100. È verosimile ipotizzare che la prevalenza sia sottostimata per la paucità o l’assenza di segni clinici nella fase iniziale della malattia e per l’assenza di un fenotipo specifico.
Malattia di Wilson: a cosa è dovuta?
Il gene coinvolto nella patogenesi della malattia di Wilson è il gene ATP7B, mappato sul cromosoma 13 alla banda q14-q21: tale gene codifica per una proteina di trasporto del rame ATP-dipendente.
Il difetto di questa proteina determina la mancata mobilizzazione del rame dai lisosomi delle cellule epatiche per essere escreto nella bile, con conseguente accumulo intraepatocitario progressivo, che determina un danno ossidativo nei mitocondri.
Anche se sono state descritta circa 400 mutazioni del gene ATP7B, nella maggior parte dei casi la malattia di Wilson è dovuta a un piccolo numero di mutazioni specifiche per quella data popolazione. Per esempio, nelle popolazioni occidentali, la mutazione del gene H1069Q è presente nel 37-63% dei casi.
Relativamente poco si sa circa l’impatto relativo delle varie mutazioni (correlazione genotipo-fenotipo), anche se la mutazione H1069Q sembra, secondo alcuni studi, portare a insorgenza soprattutto di problemi neurologici successivamente.
Malattia di Wilson: la Fisiopatologia
La condizione, come già detto, è dovuta ad un accumulo patologico del rame a livello di differenti tessuti, per alterato funzionamento della proteina codificata dal gene ATP7B.
Il rame viene assorbito nell’intestino insieme alle proteine ricche di zolfo:
- la CMT1, posta sulle cellule del piccolo intestino, trasporta il rame al loro interno, dove viene legato, in parte con qualche metallotioneinae in parte trasportato dalla ATOX1 verso l’apparato del Golgi dell’enterocita. Qui, in risposta alle concentrazioni crescenti di rame, un enzima chiamato ATP7A rilascia il rame nella vena porta e poi verso il fegato.
- le cellule epatichepossiedono anch’esse la proteina CMT1 che trasporta al loro interno il rame, dove esso viene legato da una proteina ad attività enzimatica ferrossidasica, la ceruloplasmina. Qui, l’enzima ATP7B, provvede a rilasciarlo nel sangue, oppure a rimuovere dal fegato quello in eccesso, secernendolo all’interno della bile.
Il 90-95% del rame è trasportato nel sangue legato alla ceruloplasmina, il rimanente è legato in modo labile a complessi aminoacidici o all’albumina e viene scambiato facilmente con i tessuti (il cosiddetto rame libero). Il rame viene metabolizzato dal fegato e viene escreto attraverso la bile che rappresenta la principale via di eliminazione dall’organismo.
Quando la quantità di rame all’interno del fegato è in eccesso, si verifica il danno ossidativo attraverso un processo noto come “reazione di Fenton”. Questa ossidazione patologica comporta un verificarsi di epatiti non infettive, fibrosi e cirrosi.
Il fegato rilascia nel sangue anche il rame che non è legato alla ceruloplasmina: questo rame libero si diffonde in tutto il corpo ma va a colpire soprattutto gli occhi, i reni e il cervello. Nel cervello la maggior parte del rame si deposita nei gangli basali, in particolare nel putamen e nel globo pallido. Danni a queste aree (assieme alla possibile encefalopatia epatica), sono responsabili dei sintomi neuropsichiatrici osservati nella malattia di Wilson.
Malattia di Wilso: come si manifesta?
Il fenotipo della Malattia di Wilson è molto variabile, dipendendo da molteplici fattori, tra cui l’età e il genotipo.
La patologia si manifesta tra i 5 e i 40 anni e gli esordi precoci corrispondono ad un decorso più rapido e grave.
In età pediatrica, prima dei 10 anni, la Malattia di Wilson si manifesta principalmente con un quadro di epatopatie, mentre i sintomi neuropsichiatrici sono più comuni nella tarda adolescenza, trai i 10 e i 18 anni, e nel giovane adulto.
A prescindere dall’età, i segni clinici sono frequentemente aspecifici, ad eccezione dell’anello di Kayser-Fleischer di rara osservazione in età pediatrica.
Negli adulti i sintomi che prevalgono sono quelli neurologici. Nel 10% dei pazienti la malattia di Wilson può esordire con coinvolgimento a carico di altri organi ed apparati, quali rene, occhio, scheletro e cuore.
L’eterogeneità dell’espressività clinica e laboratoristica della Malattia di Wilson spiega perché la diagnosi sia molto impegnativa e spesso la condizione misconosciuta o diagnosticata tardivamente.
- Danni epatici: I disturbi epaticipossono presentarsi con stanchezza, emorragia, dolore addominale o confusione e ipertensione portale. Inoltre, nel caso in cui la pressione nella vena porta aumenti considerevolmente, appaiono le varici esofagee, splenomegalia e ascite. Spesso nei pazienti che riportano i sintomi è diagnosticata una cirrosi epatica, con fibrosi.
Il 5% circa dei pazienti ricevono una diagnosi solo in seguito a improvvisi e acuti problemi epatici, spesso nel contesto di un’anemia emolitica . Ciò conduce a un’anomala produzione delle proteine e a difetti del metabolismo del fegato, con conseguente accumulo di prodotti di scarto, come ammoniaca, nel circolo sanguigno: il paziente quindi può sviluppare un’encefalopatia epatica.
- Sintomi neuropsichiatrici: Circa la metà dei pazienti con il morbo di Wilson presenta problemi neurologici o psichiatrici.
La maggior parte dei pazienti inizialmente manifesta un deterioramento lieve e reversibile delle capacità cognitive, assieme a cambiamenti comportamentali. In seguito giungono sintomi neurologici specifici, spesso sotto forma di parkinsonismo: aumentata rigidità, ipertono, in casi gravi camptocormia, rallentamento dei comuni movimenti, con o senza un tipico tremore alle mani a riposo, scialorrea, amimia.
Altri disturbi presenti sono: disartria, atassia e distonia temporanea.
Malattia di Wilson: sintomi neurologici
I sintomi neurologici più comuni nel morbo di Wilson sono tuttavia convulsioni ed emicrania. A volte sono presenti difficoltà a deglutire (disfagia), diplopia, affaticamento muscolare, astenia, dolore muscolare, sincope, difficoltà nella scrittura. Raramente può anche presentarsi con dei sintomi di apparente poli-neuropatia.
Problemi psichiatrici dovuti alla malattia di Wilson possono includere cambi comportamentali quali la depressione, l’ansia, la confusione mentale, fino alla psicosi e in fase avanzata sintomi somiglianti a quelli iniziali della demenza subcorticale e della demenza frontotemporale (disturbi dell’umore di origine non psichiatrica, impulsività, lentezza di pensiero, perdita di memoria). Si può infine verificare la paralisi pseudobulbare. I sintomi psichiatrici sono comunemente osservati insieme con i sintomi neurologici, raramente si manifestano da soli. Questi sintomi spesso non sono ben definiti e si possono attribuire ad altre cause. Per questo la diagnosi del morbo di Wilson è raramente fatta quando solo i sintomi psichiatrici sono presenti, in quanto il medico può essere tratto in inganno pensando a una patologia puramente psichica.
- Altri organi Diversi organi sono coinvolti dall’accumulo di rame nella malattia di Wilson:
- Occhi: caratteristici sono gli anelli di Kayser-Fleischer: essi sono dovuti alla deposizione di rame nella membrana di Descemetdella cornea. Non si verificano in tutte le persone (sono presenti nel 66% dei casi) e possono essere visibili all’esame con la lampada a fessura. La malattia di Wilson è anche associata a cataratte, pigmentazione marrone o verde della capsula;
- Renie apparato scheletrico: acidosi tubulare renale di tipo “, una malattia che porta a nefrocalcinosi, indebolimento delle ossa a causa della perdita di calcio e fosfato)con osteoporosi, osteomalacia e artrosi e occasionalmente aminoaciduria
- Cuore: la cardiomiopatiaè un problema raro, ma riconosciuto nella malattia di Wilson e può portare a insufficienza cardiaca e aritmie cardiache
- Ormonie sistema endocrino: ipoparatiroidismo, infertilità, amenorrea, pubertà ritardata e aborti spontanei abituali; raramente ipotiroidismo;
- Cute: a volte lunuledelle unghie di colore ceruleo, xerosi, pigmentazione anomala e facilità alle infezioni cutanee
- Altri sintomi: anoressia, inappetenza, perdita del gusto, secchezza e lieve alopecia, agranulocitosi, disturbi gastrointestinalie digestivi in conseguenza dei problemi al fegato, disturbi alle mucose, facilità all’anafilassi allergica.[
Malattia di Wilson: Come si diagnostica?
La malattia di Wilson deve essere sospetta in individui sopra l’anno di vita che presentino combinazioni variabili di disturbi epatici, neurologici e psichiatrici.
La diagnosi si può articolare in tre differenti momenti:
1° step: valutazione clinica per epatospenomegalia, ascite, anello di KayserFleischer; esami di laboratorio: AST/ALT, bilirubina totale/diretta, INR, ALP, ceruloplasmina sierica, cupruria delle 24 ore.
2° step: analisi molecolare (mutazioni comuni, intera sequenza genomica).
3° step: rame epatico (se analisi molecolare non conclusiva o non disponibile).
La maggior parte dei pazienti presenta test di funzionalità epatica anomali, come elevati livelli di transaminasi e bilirubina aumentata. Se il danno epatico è significativo si riscontreranno bassi livelli di albumina, PT prolungati e fosfatasi alcalina bassa.
La combinazione di sintomi neurologici, livello basso di ceruloplasmina e la presenza degli anelli di Kayser-Fleischer negli occhi sono considerati sufficienti per la diagnosi del morbo di Wilson. In molti casi, tuttavia, sono necessari ulteriori accertamenti.
Non esiste un test completamente affidabile per la diagnosi del morbo di Wilson, ma i livelli di ceruloplasmina e rame plasmatici, nonché della quantità di rame escreto nelle urine durante un periodo di 24 ore, sono utilizzati per capire la quantità di rame presente nel corpo. La prova standard ideale per la diagnosi è comunque la biopsia epatica.
Il dosaggio della ceruloplasmina è un test poco sensibile e poco specifico. Valori di ceruloplasmina 5mg/dl sono fortemente suggestivi di malattia. Il 95% dei malati e il 20% degli eterozigoti hanno valori inferiori a 20 mg/dl.
La ferritina risulta essere alta.
I valori di rame nel siero sono bassi, ma paradossalmente sono elevati nelle urine. Per l’esame, l’urina viene raccolta per 24 ore in una bottiglia con un rivestimento privo di rame. Livelli superiori a 100 µg/24h (1,6 µmol/24h) confermano la malattia di Wilson e dei livelli superiori a 40 µg/24h (0,6 µmol/24h) sono fortemente indicativi.
La biopsia epatica con dosaggio del rame su tessuto secco (indagine di secondo livello) va eseguita in quei casi in cui lo studio del metabolismo del rame, il quadro clinico ed eventualmente l’analisi genetica non sono conclusivi. Concentrazioni di rame > 250 mcg/g di tessuto secco sono da considerarsi fortemente suggestive per la diagnosi. I metodi istochimici per la rilevazione di rame sono incoerenti e inaffidabili e se effettuati da soli sono considerati insufficienti per stabilire una diagnosi precisa.
Se sono presenti sintomi neurologici, la risonanza magnetica del cervello viene di solito eseguita; essa può mostrare anche il “volto del panda gigante” caratteristico della malattia. La risonanza magnetica nucleare (RMN) e/o la tomografia assiale computerizzata (TAC) possono mostrare alterazioni nei gangli basali e nella materia bianca subcorticale, o atrofia di alcune parti di essi.
I test genetici e la ricerca delle mutazioni responsabili permettono di limitare al minimo indispensabile il dosaggio del rame su frammenti di tessuto ottenuti da biopsia epatica.
L’analisi genetica consiste nel sequenziamento dei 21 esoni del gene ATP7B e delle regioni introniche immediatamente fiancheggianti gli esoni stessi.
L’assenza di mutazioni non esclude la diagnosi: solo nell’80% dei casi è possibile giungere all’identificazione completa dei geni causali. In caso di riscontro di una mutazione non nota in letteratura, si dovranno analizzare tutti i restanti esoni del gene ed eventualmente effettuare specifici studi funzionali sulla proteina ATP7B.
Lo screening genetico è indicato per:
– confermare la diagnosi in un soggetto con clinica sospetta;
– fratelli/sorelle del soggetto affetto che ancora non hanno sintomi (possono essere sani, affetti o portatori di una mutazione);
– figli di soggetti affetti;
– partners di soggetti affetti/portatori con funzione pre-concezionale (per permettere eventuale diagnosi prenatale)
H2: Consulenza genetica
La patologia viene trasmessa con modalità autosomica recessiva. È fondamentale, pertanto, conoscere lo stato dei partner per poter fornire un rischio di ricorrenza di patologia.
Due soggetti entrambi portatori hanno un rischio di avere un figlio affetto del 25%, di avere un figlio portatore del 50% e di avere un figlio sano del 25%, per ogni gravidanza ed indipendentemente dal sesso del nascituro.
Un soggetto affetto trasmetterà la variante nel 100% alla prole, che risulterà portatrice: per conoscere il rischio di ricorrenza di patologia è necessario conoscere il genotipo del partner.
È possibile la diagnosi prenatale.
Malattia di Wilson: come si cura?
Per i soggetti affetti da Malattia di Wilson si consiglia una dieta povera di alimenti contenenti rame, quali funghi, noci, cioccolato, frutta secca, fegato animale e frutti di mare.
Sono disponibili diversi trattamenti farmacologici per la malattia di Wilson.
In generale, la penicillamina è il primo farmaco usato: questa lega il rame (chelazione) e lo porta a essere escreto attraverso le urine. La penicillamina non è priva di controindicazioni: circa il 20%, infatti, soffre di effetti collaterali o complicanze del trattamento come il lupus farmaco-indotto (causa dolori articolari ed eruzioni cutanee). In coloro che hanno anche sintomi neurologici, la quasi metà soffre di un paradossale peggioramento dei sintomi
I pazienti intolleranti alla penicillamina possono incominciare la terapia con cloridrato di trientina, che possiede anche proprietà chelanti. Alcuni raccomandano la trientina come trattamento di prima linea, ma l’esperienza con penicillamina è maggiore. Un altro agente con nota attivitàchelante è il tetratiomolibdate: questo trattamento però è ancora considerato sperimentale, anche se alcuni studi hanno mostrato un effetto benefico.
Una volta che tutti i valori sono ritornati alla normalità, lo zinco (di solito sotto forma di una prescrizione di acetato di zinco, chiamato GALZIN) può essere utilizzato al posto di chelanti per mantenere stabili i livelli di rame nel corpo.
Nei rari casi in cui nessuno dei trattamenti per via orale risulti efficace e soprattutto nei casi di grave malattia neurologica, il dimercaprolo risulta necessario.
Le persone che sono asintomatiche vengono generalmente trattate pure, poiché l’accumulo di rame può causare danni nel lungo termine. Non è chiaro se queste persone possano essere trattate meglio con la penicillamina o con l’acetato di zinco.
Il trapianto di fegato è una cura efficace per la malattia di Wilson, ma è utilizzato solo in condizoni particolare gravità a causa dei numerosi rischi e complicazioni associate alla procedura chirurgica. Viene utilizzato principalmente nelle persone con insufficienza epatica fulminante che non rispondono al trattamento medico o in quelli con avanzata malattia epatica cronica. Il trapianto di fegato è evitato nei casi di grave malattia neuropsichiatrica, in cui la sua efficacia non è stata dimostrata.
Malattia di Wilson: associazione pazienti
- Associazione nazionale Malattia di Wilson malattiadiwilson.it. Per informazioni: info@malattiadiwilson.it
Bibliografia
- PDTA Regione Lombardia Malattia di Wilson – https://malattierare.marionegri.it/images/PDTA/Schede/wilson.pdf
- Ala A et al, Wilson’s Disease, The Lancet, 2007 – https://pubmed.ncbi.nlm.nih.gov/17276780/
- Tanzi RE et al, The Wilson disease gene is a copper transporting ATPase with omology to the Menkes disease gene, Nature Genetics, 1993 – https://pubmed.ncbi.nlm.nih.gov/8298641/
- Brewer G. et al, Wilson’s disease: clinical management and therapy, Journal of Hepatology, 2004 – https://www.journal-of-hepatology.eu/article/S0168-8278(04)00531-8/fulltext
- Das S et al, Wilson’s disease: an update, Nature Reviews Neurology, 2006 – https://pubmed.ncbi.nlm.nih.gov/16932613/
- Czlonkowska A et al, Wilson dieseas, Nat Rev Dis Primers, 2018 – https://pubmed.ncbi.nlm.nih.gov/30190489/
- Mulligan C et al, Wilson disease: an overview and approach to management, Neurol Clin, 2020 – https://pubmed.ncbi.nlm.nih.gov/32279718/
- Schilsky Mi, Wilson disease: diagnosis, treatment and follow-up, Clin Liver Dis, 2017 – https://pubmed.ncbi.nlm.nih.gov/28987261/





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}