Caratteristiche e aspetti genetici
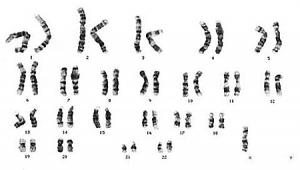
L’assetto cromosomico diploide prevede la presenza di 22 coppie di autosomi e di una coppia di cromosomi sessuali che sarà, quest’ultima, XX in un soggetto di sesso femminile e XY in un soggetto di sesso maschile.
La presenza di una anomalia cromosomica quale la monosomia del cromosoma X configura una condizione clinica denominata sindrome di Turner: in questo caso l’assetto cromosomico sarà quindi 45,X. Esistono altre anomalie cromosomiche associate alla suddetta sindrome: la monosomia X infatti rappresenta il 50% dei casi, nel 25% si riscontra la presenza di un mosaicismo cromosomico (il cariotipo 46XX/45,X è, tra questi casi, la forma più frequente) e nel restante 25% si riscontra un cromosoma X strutturalmente anomalo (ad esempio una delezione X, l’isocromosoma X, o il cromosoma dicentrico) che può presentarsi in forma completa o a mosaico.
Nei casi di evidenza citogenetica di un mosaicismo coinvolgente il cromosoma Y sarà importante valutare le gonadi per le possibili complicanze degenerative influenzate dalla produzione di testosterone. Qualora la presenza del cromosoma Y non sia evidente al cariotipo è importante escludere un eventuale possibile basso mosaicismo mediante l’utilizzo di tecniche di indagine molecolare.
La monosomia del cromosoma X presenta un’alta incidenza di selezione naturale e rappresenta infatti una delle anomalie cromosomiche di più frequente riscontro in seguito ad analisi del cariotipo su materiale abortivo.
Caratterizzare l’assetto cromosomico fetale, in seguito a un aborto spontaneo, è fondamentale: tale procedura infatti serve a individuare i casi in cui le cause di abortività o di complicanze gravidiche, come recentemente descritto in letteratura, siano legati ad alterazioni cromosomiche, come ad esempio quelle a carico del cromosoma X, che potrebbero essere di origine materna e modificare pertanto il rischio di ricorrenza.

Sindrome di Turner e gravidanza: la diagnosi prenatale
In alcuni casi, sin dal I trimestre di gravidanza, è possibile porre diagnosi prenatale di sindrome di Turner. Nella letteratura internazionale è ben consolidata la conoscenza che la presenza di alcuni soft markers ecografici, quali ad esempio l’aumento della translucenza nucale, l’idrope fetale, le anomalie dell’accrescimento fetale, o la presenza di anomalie malformative fetali possano rappresentare un indicatore importante per avviare indagini prenatali invasive o non invasive.
La diagnosi di sindrome di Turner può anche essere effettuata in seguito al ricorso a indagini prenatali non invasive quali quelle che si basano sullo studio del DNA fetale circolante (NIPT): studi recenti sottolineano che queste tecnologie, pur non essendo ancora considerate diagnostiche in particolare per le anomalie a carico dei cromosomi sessuali, possono fornire una indicazione per successivi approfondimenti genetici.
Diagnosi in epoca pediatrica
Alla nascita possono presentarsi dei segni clinici evocativi per sindrome di Turner e tra questi l’edema del dorso dei piedi, lo pterigio del collo o la presenza di cardiopatie congenite quali la coartazione aortica spesso non evidenziabili ecograficamente in epoca prenatale.
Una manifestazione clinica presente nella quasi totalità dei casi è la bassa statura per cui, a volte, la diagnosi giunge nel corso di indagini genetiche volte a comprendere il perché una bambina non cresce in modo adeguato all’età.
Recentemente sono state aggiornate e pubblicate le indicazioni per il management della sindrome di Turner dove vengono indicati i percorsi assistenziali da seguire in relazione all’età del soggetto.
Diagnosi in epoca puberale
Una caratteristica comune nei soggetti con sindrome è di Turner è l’assenza del menarca per cui è possibile che si giunga alla diagnosi di tale anomalia cromosomica nel corso di indagini volte a comprendere il perché non sia comparso il ciclo mestruale all’età prevista.
In questi casi la valutazione ecografica ginecologica farà emergere, generalmente, la presenza di una ipoplasia ovarica e uterina dovuta a una ridotta produzione di estrogeni (alterazione dell’asse ipotalamo-ipofisi-ovaio) legata, a sua volta, all’assenza del cromosoma X o alle anomalie strutturali del cromosoma X responsabili della condizione.
In epoca prepuberale, da alcuni anni, è possibile utilizzare dei protocolli di terapia su base estrogenica che hanno ottenuto dei risultati significativi sia sull’accrescimento staturale che in generale sulla funzione ormonale.
Diagnosi in età adulta
Legata all’alterata produzione ormonale vi è anche la possibilità che i soggetti con sindrome di Turner, generalmente quelli con assetto cromosomico a mosaico, non diagnosticati in epoca antecedente e non trattati con adeguata terapia, possano presentare un ciclo irregolare, infertilità e in rari casi menopausa precoce.
Pertanto l’esecuzione del cariotipo in donne che presentano tali caratteristiche risulta fondamentale per individuare la causa della problematica riproduttiva e per valutare terapie appropriate e stimare il rischio di ricorrenza nella prole di anomalie dei cromosomi sessuali.
Anche in questi casi risulta essenziale seguire le indicazioni per il management e i dei protocolli di terapia per la sindrome di Turner che sono validati a livello internazionale.
Conclusioni
La sindrome di Turner è una condizione genetica complessa che può essere diagnosticata in epoca prenatale o durante differenti fasi della vita.
La possibilità di utilizzare delle terapie ormonali in grado di agire sull’asse ipotalamo-ipofisi-ovaio ha cambiato nel tempo la storia naturale di questa condizione aprendo scenari di cura che in passato non erano possibili.
La consulenza genetica e l’inserimento di questi soggetti in percorsi di follow up multidisciplinari può permettere di programmare i corretti interventi terapeutici atti a migliorare la qualità di vita nonché la gran parte delle caratteristiche proprie della sindrome di Turner, ivi compresa, in alcuni casi, la possibilità di migliorare l’aspetto della fertilità attraverso l’utilizzo di tecniche di PMA.
Non va infine trascurato l’aspetto di supporto svolto dalle Associazioni di Pazienti e Familiari che possono fornire indicazioni sui Centri di riferimento a cui rivolgersi nonchè favorire lo scambio di informazioni.
Per ulteriori approfondimenti puoi contattare i genetisti di Bgenetica o, qualora dovessi rientrare in una delle condizioni prima illustrate, valutare la possibilità di eseguire una consulenza genetica.
Bibliografia:
1) Mardy AH et al. Maternal Genetic Disorders and Fetal Development. Prenat Diagn Feb 3, 2020
2) Wiechec M et al. What Are the Most Common First-Trimester Ultrasound Findings in Cases of Turner Syndrome? J Matern Fetal Neonatal Med 2017 Jul;30(13):1632-1636
3) Wang Y eta al. Cell-free DNA Screening for Sex Chromosome Aneuploidies by Non-Invasive Prenatal Testing in Maternal Plasma. Mol Cytogenet 2020 Mar 12;13:10
4) Gravolth CH et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017 Sep;177(3):G1-G70
5) Hasegawa Y et al. Ultra-low-dose Estrogen Therapy for Female Hypogonadism. Clin Pediatr Endocrinol 2020;29(2):49-53
6) Ye M et al. Progress in Fertility Preservation Strategies in Turner Syndrome. Front Med (Lausanne) 2020 Jan 24;7:3
7) Gravolth CH et al Turner Syndrome: Mechanisms and Management. Nat Rev Endocrinol 2019 Oct;15(10):601-614




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}